A team led by Prof. JIANG Bin from the University of Science and Technology of China (USTC) made a series of breakthroughs in chemical dynamics simulations of molecular, condensed phase and interfacial systems by applying an atomistic neural network (AtNN). A review of their works was published in WIREs Computational Molecular Science on November 16th.
AtNN has been widely used in physical chemistry research, particularly in chemical dynamics simulations. By decomposing the system properties into the contribution of each atom, AtNN can satisfy different symmetries and periodicities of molecular, condensed phases, and interfacial systems, thereby achieving accurate and efficient molecular dynamics simulations of complex systems.
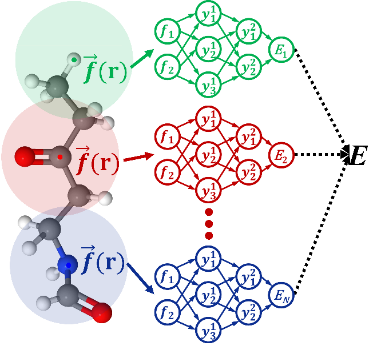
The framework of AtNN. (Image by ZHANG Yaolong et al.)
Prof. JIANG Bin’s team has published a series of papers on using AtNN to predict potential energy surface and chemical properties. Based on their previous research, this review introduced the basic concepts and the physical ideas of the AtNN methods and discussed various strategies to improve the efficiency of the atomic environment description in different AtNN methods. The embedded atomic neural network (EANN) proposed by Prof. JIANG Bin’s team uses the square of the linear combination of Gaussian-type orbitals (GTOs) to calculate the three-body correlation, significantly improving the efficiency. In terms of description, EANN combines multiple radial functions to enhance the two-body correlation. Furthermore, by using a message passing network to recursively incorporate non-local interactions outside the atomic environment, the accuracy of the model is significantly improved compared to AtNN, which is based entirely on local multi-body descriptors.
In addition, the paper also summarized recent works on generalizing the scalar AtNN models to represent tensorial quantities. These works mainly focus on tensorizing the permutation-invariant output of the AtNN to satisfy the rotational invariant symmetry of tensors. For example, by using coordinates and gradients to introduce directional properties and construct first-order or second-order tensors to represent response and transition properties like dipole moment and polarizability. Great progress has also been made in describing more complex tensors like electronic Hamiltonian with AtNN.
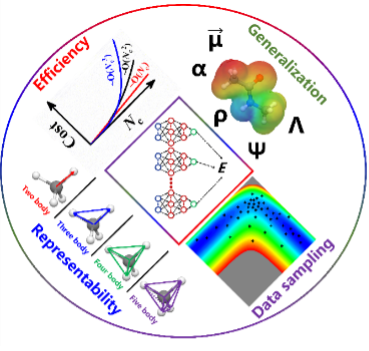
The main focus of the AtNN. (Image by ZHANG Yaolong et al.)
The training data set is crucial to the model construction of a specific system in AtNN. The active learning algorithms like error search developed by Prof. JIANG Bin’s team, combined with molecular dynamics trajectories, are able to search for areas with large uncertainty in the model and sample new configurations to add to training set, which can improve the model training. Another challenge in data sampling is to efficiently consider the effect of atomic substitution on configuration similarity. The efficiency of AtNN in solving the above problems was proved by several examples of its application in gas-phase surface systems.
In the future, the team expects to generalize AtNN methods to express long-range interactions, chemical properties under external fields, and solve the Schrödinger equations of electrons and nuclei.
Paper link: https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1645
(Written by ZHENG Ruichen, edited by ZHANG Yihang, USTC News Center)